Нашёл!
Для всякого бесконечно малого процесса энтропия, вообще говоря, удовлетворяет неравенству:
dS \geq \frac{\delta Q}{T}.
Так как в дальнейшем все процессы будем считать изотермическими, то, используя свободную энергию Гельмгольца \Psi=U-TS, продифференцирую его при постоянной температуре, т.е. d\Psi_T=dU-TdS. Выражая первый закон термодинамики \delta Q = dU + \delta A, получу:
d\Psi_T≤-\delta A,
иначе говоря, внешняя работа -\delta A над системой, вообще говоря, больше изменения свободной энергии. Если система изолирована, то \delta A=0, а значит d\Psi_T\leq0, т.е. в термодинамически стабильном состоянии свободная энергия \Psi минимальна.
То же самое происходит и с термодинамическим потенциалом Гиббса \Phi=\Psi+PV. Так как d\Phi=d\Psi+PdV+VdP=d\Psi+\delta A+VdP, то при постоянной температуре и с учётом сказанного выше получаю
d\Phi\leq-VdP.
Теперь потихоньку будем подходить к самой задаче. Так как речь идёт о процессе на границе раздела жидкости с газом, то механическая устойчивость требует того, чтобы dP=0, то есть опять же получается, что \Phi стремится к минимуму.
Для равновесии двух фаз нужно, чтобы потенциалы Гиббса были равны. Рассмотрим систему из двух фаз с массами m_1 и m_2. Потенциал \Phi в сумме равен
\Phi=m_1\varphi_1+m_2\varphi_2,
где \varphi_1 и \varphi_2 являются удельными термодинамическими потенциалами. Массы могут превращаться в друг друга, но полная масса системы неизменна, а значит dm_1=-dm_2. Кроме того, происходящие процессы должны быть направлены в сторону уменьшения \Phi. Если \varphi_1>\varphi_2, то для минимума \Phi вся фаза 1 должна превратиться в фазу 2, и наоборот. Только в случае
\varphi_1(P,T)=\varphi_2(P,T)
возможно фазовое равновесие. Оттуда и до вывода формулы Клапейрона-Клаузиуса недалеко)
Отмечу, что
\left (\frac{\partial \Phi}{\partial P}\right)_T=V.
Для удельного потенциала V будет являться удельным объёмом вещества v. Рассмотрим жидкость под давлением P_{liq} в равновесии с её паром с давлением P_{vap}. Так как для двух фаз \varphi_{liq}=\varphi_{vap}, то рассмотрим их малые изменения при постоянной температуре. Получим
\left (\frac{\partial \varphi_{liq}}{\partial P_{liq}}\right)_T dP_{liq}=\left (\frac{\partial \varphi_{vap}}{\partial P_{vap}}\right)_T dP_{vap},
или же
v_{liq}dP_{liq}=v_{vap}dP_{vap}.
Теперь введу предположение, что пар - идеальный газ. Тогда v_{vap}=1/\rho_{vap}=(RT)/(\mu P_{vap}), и
v_{liq} dP_{liq}=\frac{RT}{\mu} \frac{dP_{vap}}{P_{vap}}.
Проинтегрирую это выражение и избавлюсь от никому ненужных индексов:
\Delta P = \frac{\rho RT}{\mu} \ln \frac{P}{P_0},
и, наконец-то (!):
P=P_0\exp \frac{μ\Delta P}{ρRT}.
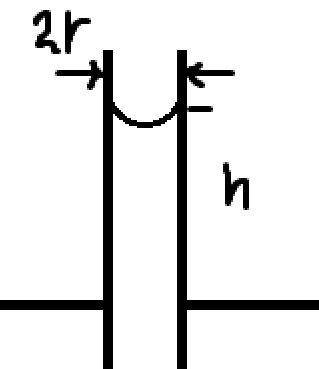
Осталось положить \Delta P = 2\sigma/r.