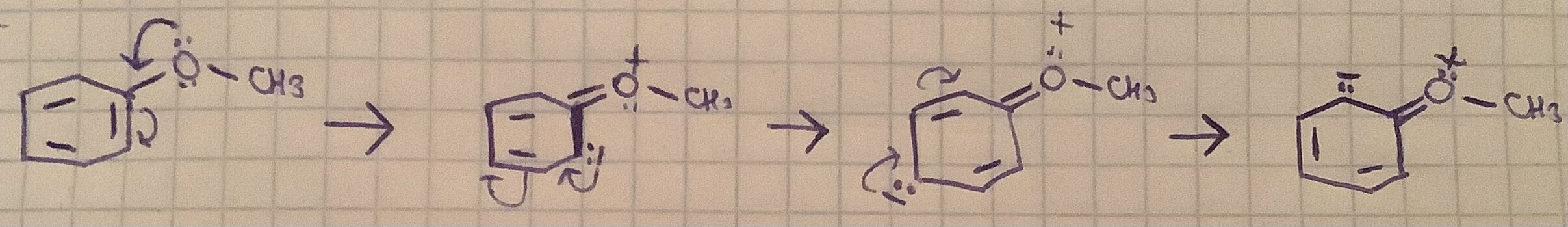
Нарисовал резонансные структуры, и видно, что среди о,m,p самые деэкранированные - углероды в m позиций, т.к у них скамит электроны кислород + в резонансных структурах на них ни разу не падал отрицательный заряд, с этим я согласен.
В случае с о и p, вроде в резонансных структурах они одинаково несут на себе отрицательный заряд, но о-углероды ближе к электроотрицательному кислороду, поэтому должны быть беднее.
У меня что-то сам по себе возник ответ, однако я не могу быть точно в нём уверен, может дело в том, что в 50% структур отрицательный заряд находится на о-углеродах, в то время как на р-углеродах только в 25% случаев, это и делает о-углероды такими богатыми на
электроны, несмотря на то, что кислород очень электроотрицательный. Может есть другие причины?
Как понять, что вы даже не открывали ссылку, которую я скидывал раньше
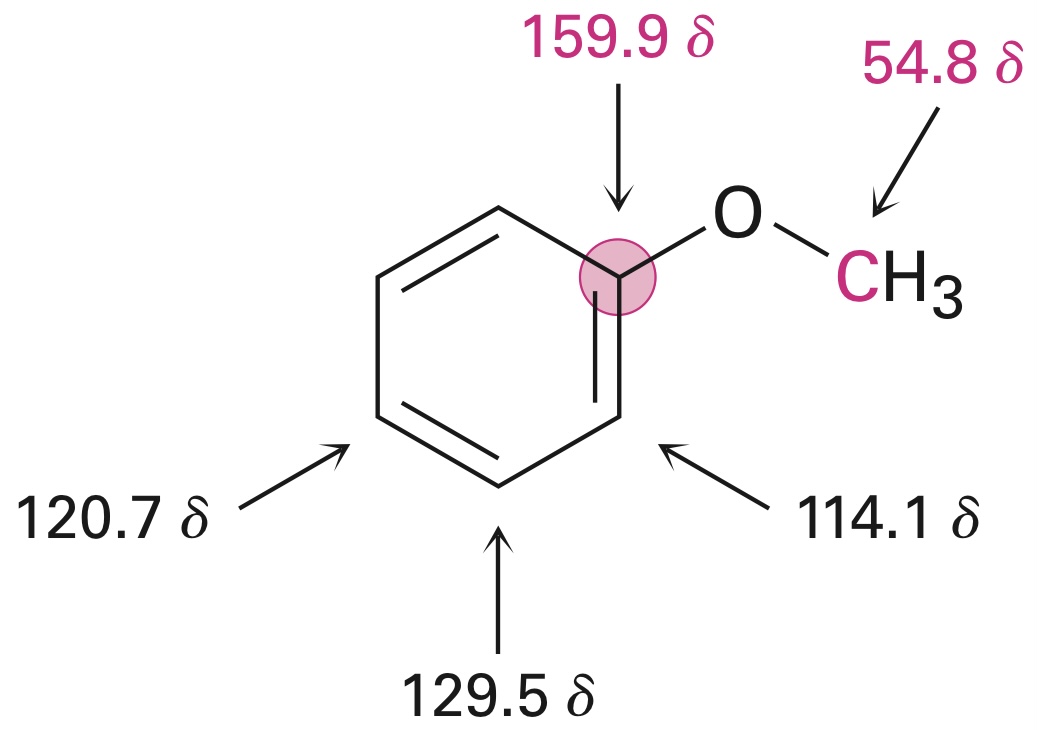
[In] the reported 1H NMR spectrum for nitrobenzene […] the ortho protons are the most deshielded, followed by the para, and, finally, the meta. This makes sense when rationalizing the inductive effect of the nitro group by resonance, which makes the ortho and para carbons deficient in electrons, hence deshielding them. Furthermore, the ortho protons are closer to the nitro group, hence they are the most deshielded of all, so the from more to less deshielded: ortho > para > meta.
However, [in] the reported 13C NMR spectrum […] the positions are now ipso > para > meta > ortho. Why is the trend so different for the carbon-13 NMR?
Да, но в каждой структуре он находится на разных о-углеродах. Иными словами 50% структур ставят заряд на о-, но в каждой только на одном о-, а о- у нас два, значит итого на каждый о-углерод так же приходится по 25%.
Неправда. Длинные и непонятные формулы были после словесного аргумента.
As I wrote in the comments, there is not really a good way of arriving at these conclusions for a general aromatic molecule. The energy differences at play in these systems are so small that qualitative arguments will often be insufficient to get the order right. The diamagnetic term, which can be qualitatively rationalized by arguments based on electron density, for non-H atoms, is dominated by the paramagnetic term, which is, in some sense, an orbital response term due to the application of the magnetic field of NMR.
Дальше он три параграфа посвящает объяснению парамагнитного эффекта без формул.