Изучение химических сэндвичей
Авогадро можно использовать для рассмотрения \pi-\pi взаимодействий.
Для начала возьмем экспериментальную геометрию бензола. Если вы сохраните следующие данные в файле c расширением .xyz[1], например benzene.xyz, вы можете открыть этот xyz файл в Авогадро и увидеть привычную гаечку.
12
Experimental geometry for benzene
C 0.0000 1.3970 0.0000
C 1.2098 0.6985 0.0000
C 1.2098 -0.6985 0.0000
C 0.0000 -1.3970 0.0000
C -1.2098 -0.6985 0.0000
C -1.2098 0.6985 0.0000
H 0.0000 2.4810 0.0000
H 2.1486 1.2405 0.0000
H 2.1486 -1.2405 0.0000
H 0.0000 -2.4810 0.0000
H -2.1486 -1.2405 0.0000
H -2.1486 1.2405 0.0000
Если открыть этот файл в Авогадро и запустить оптимизацию структуры в MMF94, геометрия не изменится, но мы можем зафиксировать значение энергии: \pu{E= 67.9391 kJ mol-1}.

Теперь сделаем наш сэндвич: расположим еще одну молекулу бензола прям над первой на расстоянии 4.5 \AA.
24
Experimental geometry for benzene
C 0.0000 1.3970 0.0000
C 1.2098 0.6985 0.0000
C 1.2098 -0.6985 0.0000
C 0.0000 -1.3970 0.0000
C -1.2098 -0.6985 0.0000
C -1.2098 0.6985 0.0000
H 0.0000 2.4810 0.0000
H 2.1486 1.2405 0.0000
H 2.1486 -1.2405 0.0000
H 0.0000 -2.4810 0.0000
H -2.1486 -1.2405 0.0000
H -2.1486 1.2405 0.0000
C 0.0000 1.3970 4.5000
C 1.2098 0.6985 4.5000
C 1.2098 -0.6985 4.5000
C 0.0000 -1.3970 4.5000
C -1.2098 -0.6985 4.5000
C -1.2098 0.6985 4.5000
H 0.0000 2.4810 4.5000
H 2.1486 1.2405 4.5000
H 2.1486 -1.2405 4.5000
H 0.0000 -2.4810 4.5000
H -2.1486 -1.2405 4.5000
H -2.1486 1.2405 4.5000
Откроем в Авогадро. Сэндвич выглядит так (я включил отображение Labels в Display Types)
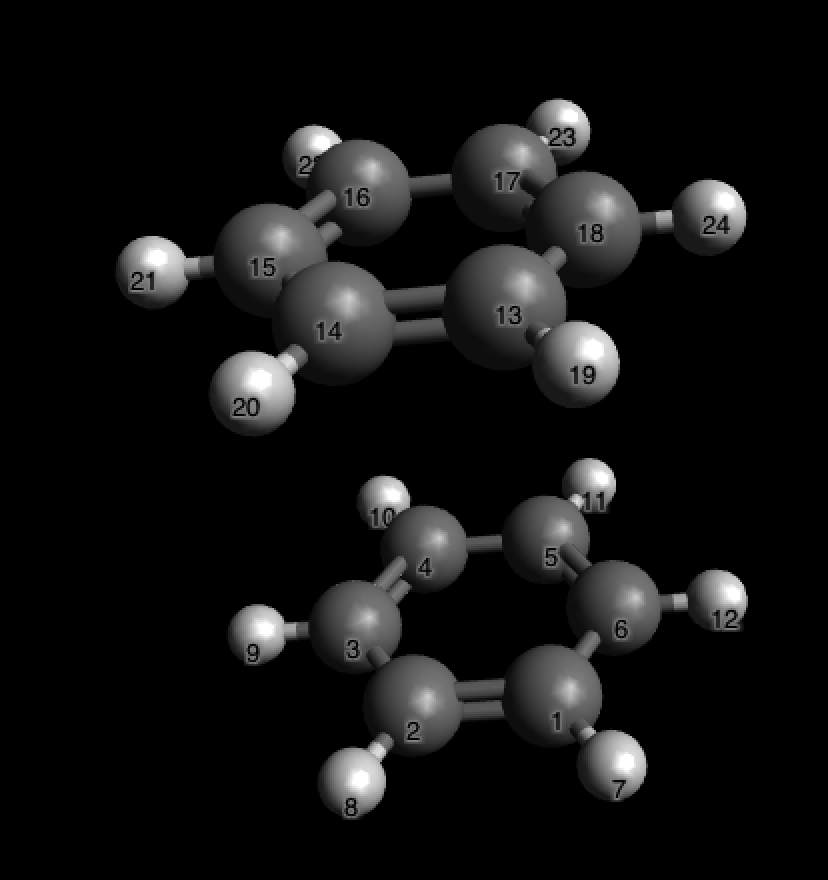
Давайте замерим расстояние между атомом углерода 1 и 13. Для этого выберем инструмент линейки и нажмем на эти два атома по очереди:
Расстояние 4.5 \AA, как мы и хотели:

Прежде чем запустим оптимизацию структуры, надо установить некоторые ограничения, чтобы одна молекула бензола оставалась параллельна второй. Для этого идем в Extensions → Molecular Mechanics → Constraints…
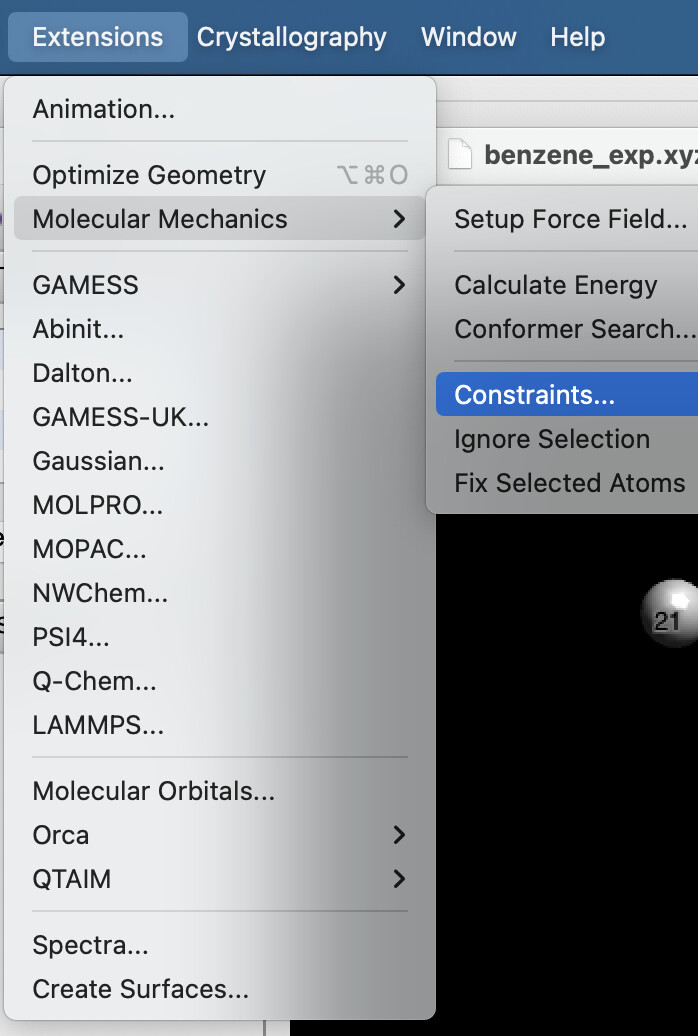
И выставляем Fix atom X и Fix atom Y по очереди для атомов 1 и 13 (т.е. мы фиксируем их x и y координаты, они не могут меняться при оптимизации):
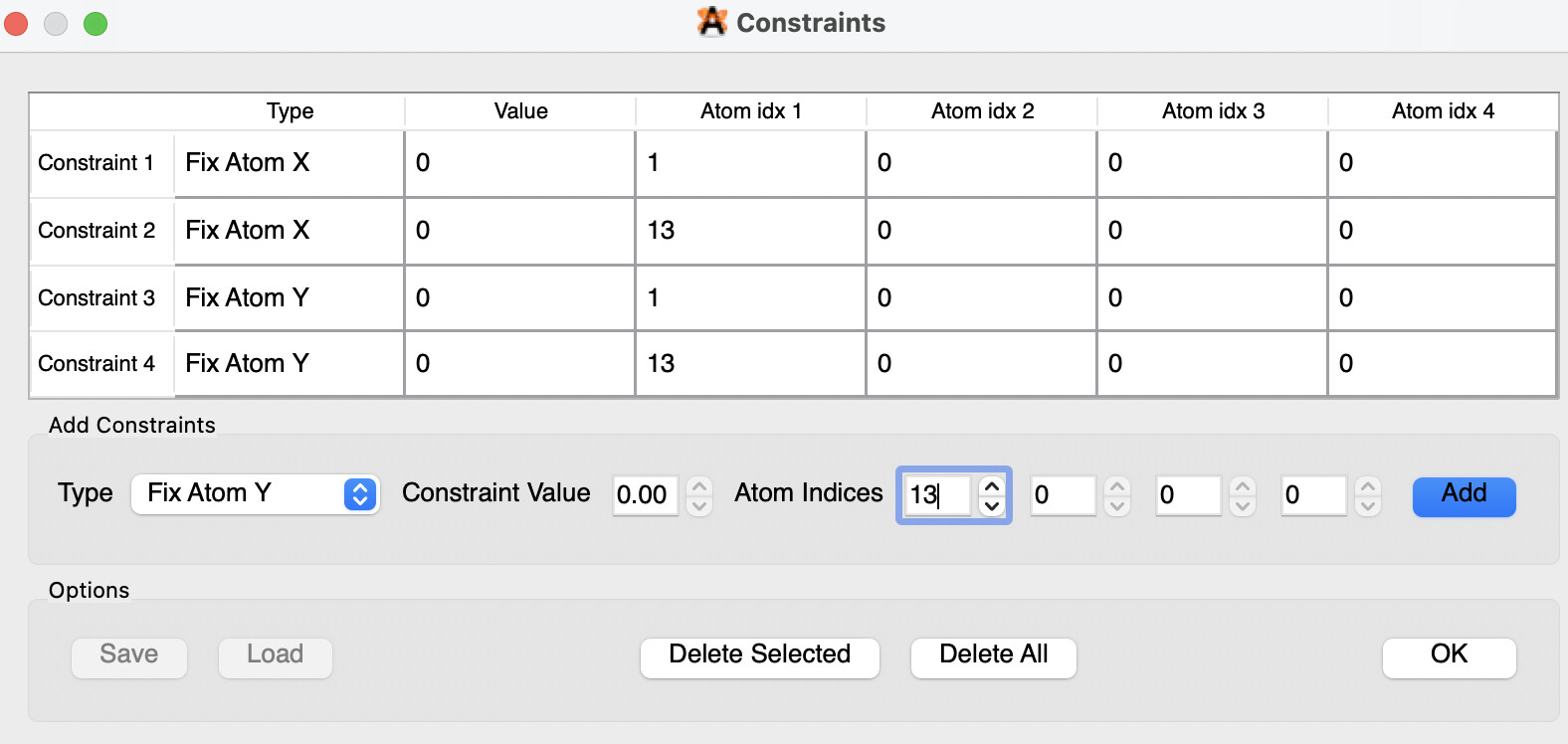
Жмем ОК и запускаем оптимизацию с MMF94. Если вы все сделали правильно, вы увидите Constraints: 4 и через минуту (или две) оптимизация завершится:
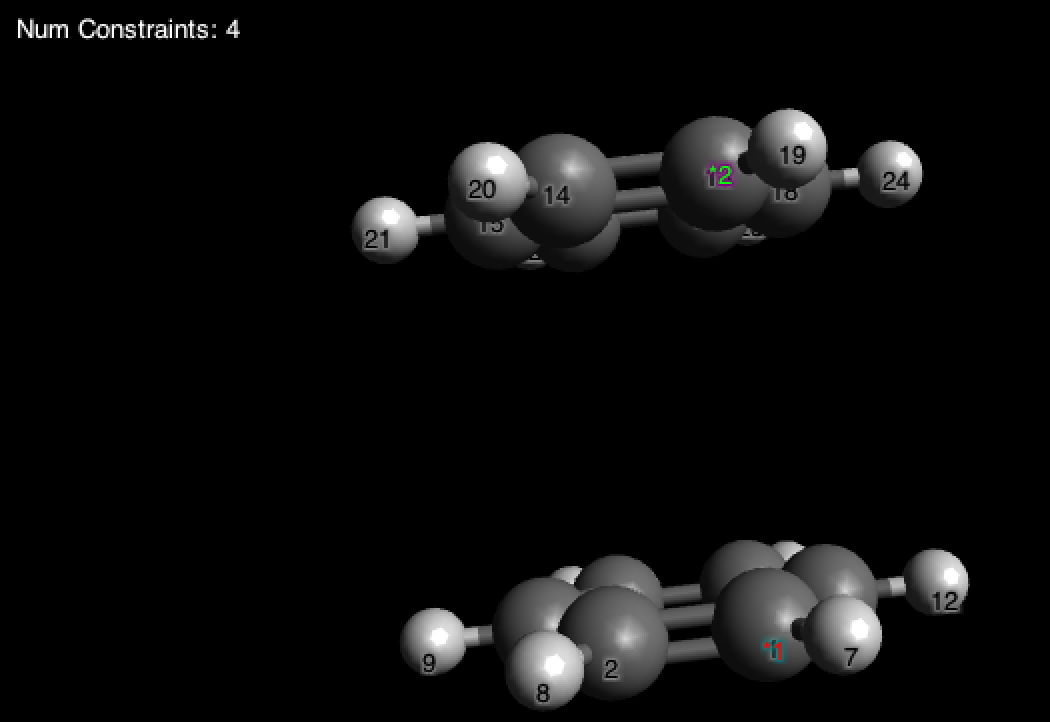
И тут у нас снова тот редкий случай, когда мы можем хоть что-то сделать с показателями энергии. Посмотрите на значение энергии, который получился в результате оптимизации сэндвича. Отнимите от этого значения две энергии бензола (\pu{E= 67.9391 kJ mol-1}, как было найдено выше) и вы получите энергию \pi-\pi взаимодействия.
Более точными квантовохимическими расчетами[2] можно получить расстояние между молекулами бензола в 3.9 \AA и энергию взаимодействия в районе \pu{7.32 kJ mol-1}. Насколько результаты MMF94 близки к этим числам? Повторите расчеты с силовым полем GAFF.
xyzфайлы имеют следующую структуру: на первой строчке указано количество атомов (N) в молекуле, вторая строчка для комментариев, а последующие N строчек указывают вид атома и его расположение в трехмерном пространстве: координаты (x,y,z) в \AA. 1\AA = \pu{10^-10 m} ↩︎CCSD(T) в complete basis set limit ↩︎