Введение
При изучении химии нередко возникают ситуации, в которых необходимо хорошо представлять как выглядит молекула в трехмерном пространстве. Например, если мы хотим показать метилциклогексан в аксиальной конформации, мы его нарисуем так:

Но из такого изображения далеко не всегда понятно в чем собственно проблема аксиальной конформации, или, иными словами, откуда берется то самое 1,3-аксиальное стерическое отталкивание?
Другой пример: бициклобутан. Скелетная структура выглядит очень странно и можно задаться вопросом: а как такая (центральная) связь вообще может существовать? Ведь эта связь должна быть в 1.4 раза длиннее обычных С-С связей!

Для разрешения таких вопросов и более наглядного изучения химии можно пользоваться программой Avogadro.
Что такое Avogadro?
Прежде чем мы начнем использовать эту программу, стоит понять для чего она была создана: это программа для молекулярного моделирования, написанная в основном для использования в научных исследованиях. Например, для визуализации данных, полученных в результате квантовохимических вычислений[1].
Иными словами, программа написана для исследователей, не для школьников, не для учителей, а значит она не обязана быть понятной и абсолютно интуитивной. Более того, наверное 80% функционала просто-напросто недоступны без результатов квантхим вычислений. И тем не менее, за оставшимися 20% этой программой стоит пользоваться в образовательных целях.
Установка Avogadro
Заходим на сайт программы и жмем Download.. Должна скачаться версия 1.2.0.[2] Еще один нюанс: эта версия была выпущена в 2016 году, а значит, неизбежно, ряд вещей может уже не работать.[3]
Скачиваем, устанавливаем, радуемся.
Для чего можно использовать Avogadro?
Строим модели молекул
Поскольку лучше один раз увидеть, чем сто раз услышать, просто посмотрите видео:

Находим оптимальную геометрию конформаций
Вот скажем вы нарисовали циклогексан, но получился он не очень симметричным:

Жмем на иконку с буквой Е (auto optimization tool), выбираем Force field: MMF94 и жмем Start
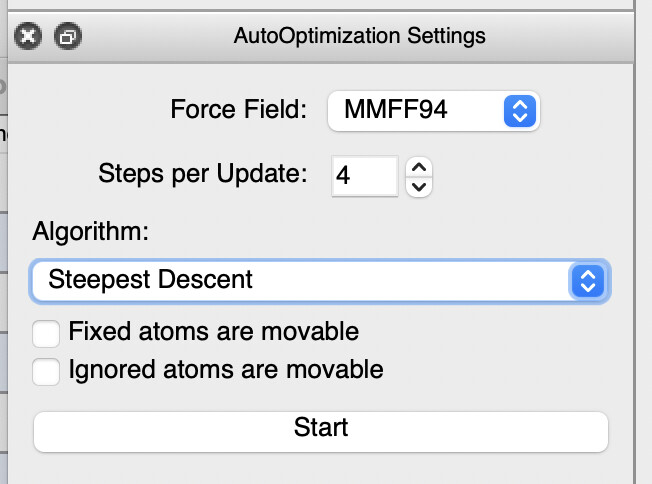
Через некоторое время вы можете наслаждаться прекрасной конформацией кресла:
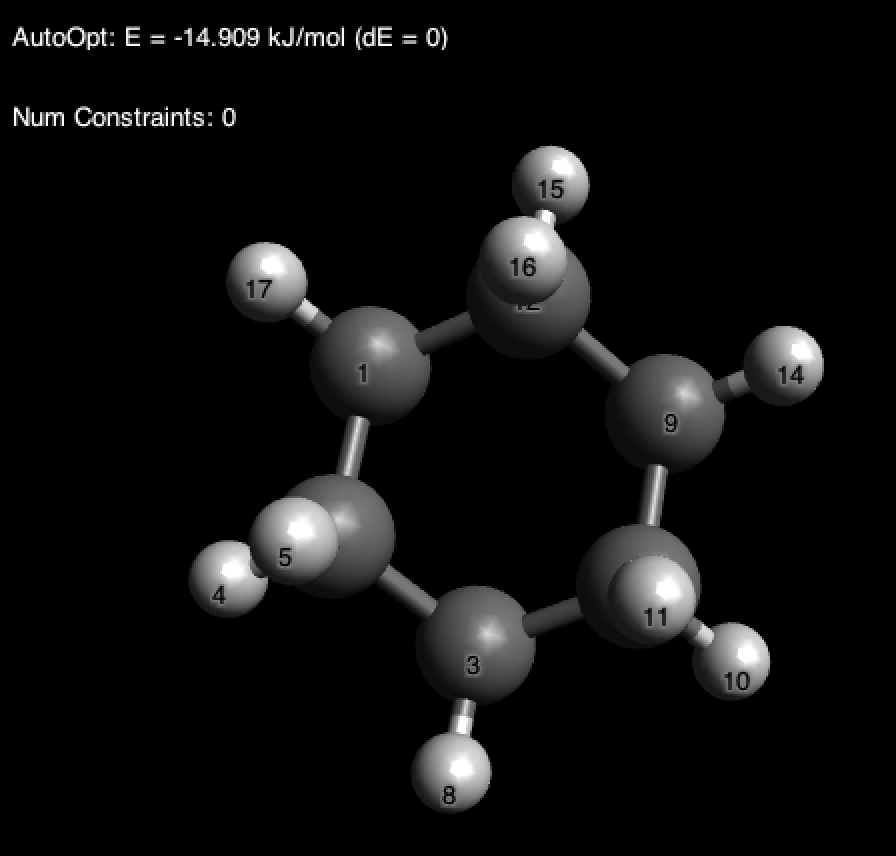
У вас сразу может возникнуть вопрос: о, а там показывают энергию, что это значит? Ровным счетом ничего.[4] Обращайте внимание только на полученную геометрию.
Давайте попробуем добавить метильную группу в аксиальное положение и заново оптимизируем структуру:

И теперь у нас почти все готово для того чтобы понять природу 1,3-стерического отталкивания. Осталось только открыть окошо Display Types:
И поставить галочку возле Van der Waals Spheres (нажав на гаечный ключ можно регулировать прозрачность сфер):

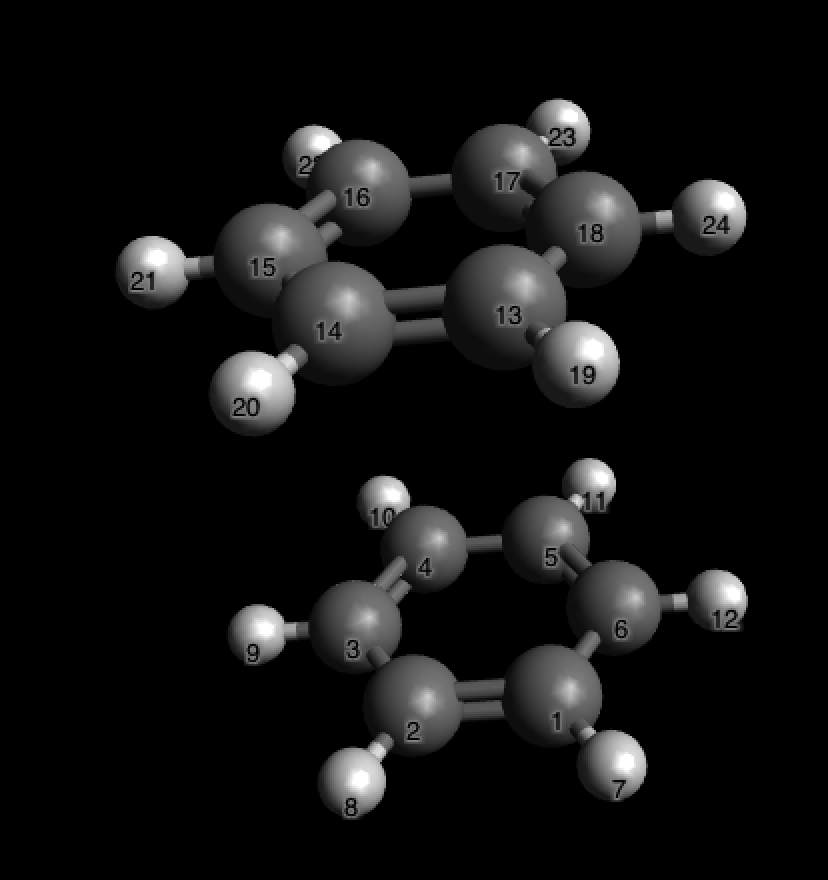
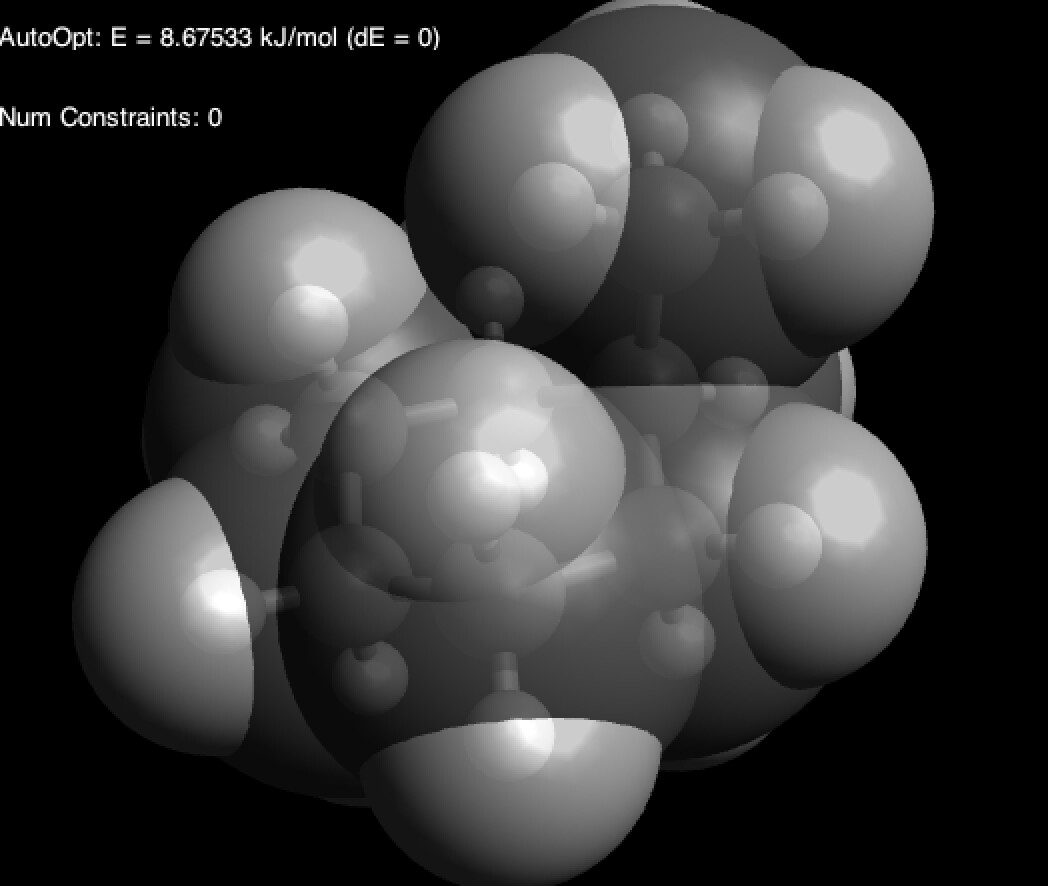
Тада! Мы видим, что атомы водорода метильной группы и аксиальные атомы водорода гораздо ближе друг к другу, чем кажется:
Дальнейшие шаги
В целом, в 99% случаев Авогадро пригодится вам именно как программа для поиска стабильной геометрии и визуализации молекул. Как вы могли догадаться, есть 1% не показанных выше возможностей. С ними можно ознакомиться в дополнительных сообщениях к этой теме:
В отдельном сообщении рассказано о том что такое силовые поля и как авогадро находит оптимальные геометрии.
Пионером квантхим вычислений была программа Gaussian, но сопровождающий софт для визуализации GaussView продается за отдельную плату ($750 за одну лицензию для академического использования). Понятное дело, что платить такие деньги крайне неприятно и в таких условиях рождаются открытые open source проекты как Avogadro. ↩︎
Вы можете наткнуться на т.н. Avogadro2, которая находится в бета версии и имеет нумерацию 1.97. Если коротко – по состоянию на август 2022 г. эту версию можно смело игнорировать. Это сплошное разочарование, никаких положительных эмоций она у меня не вызывает. ↩︎
Например на macOS еще со времен Catalina перестала работать визуализация МО, но для вас это не проблема – для МО нужны квантхим расчеты. ↩︎
Абсолютные значения этой энергии абсолютно бессмысленны. В очень редких случаях ими можно пользоваться в сравнительном ключе. ↩︎