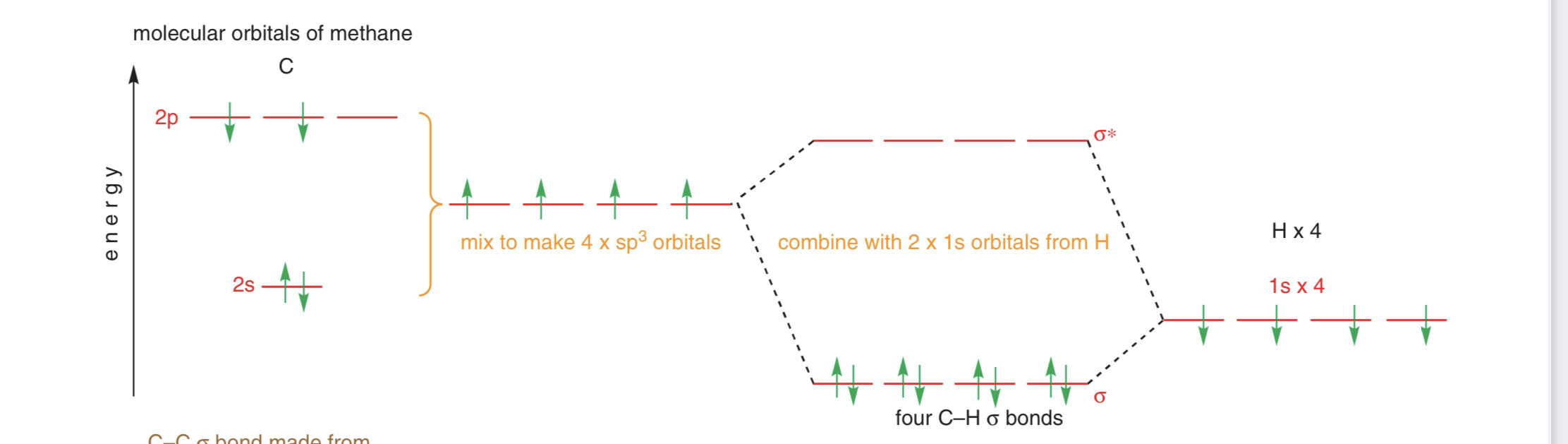
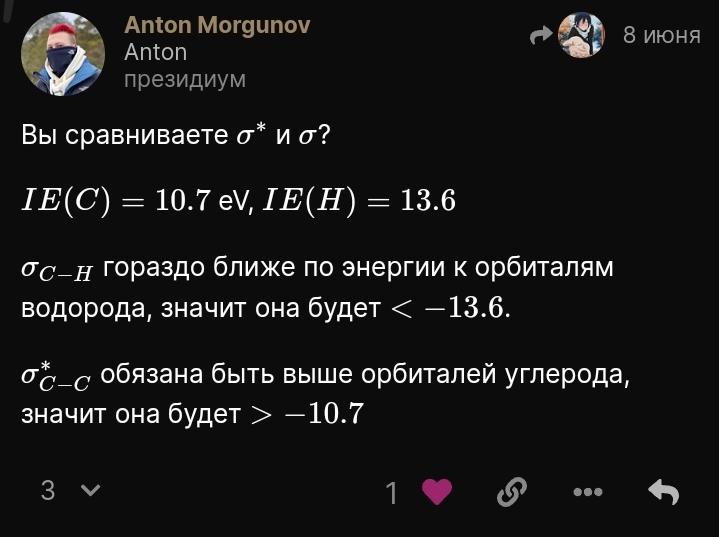
Тут показали диаграмму МО карбонильной группы (формальдегида). А дальше в книге говорят так:
Look at the filled π orbital in the MO energy level diagram. It is more similar in energy to the p orbital on O than the p orbital on C. We can interpret this by saying that it receives a greater contribution from the p orbital on O than from the p orbital on C. Consequently the orbital is distorted so that it is bigger at the O end than at the C end, and the electrons spend more time close to O. The same is true for the σ bond, and the consequent polarization of the C=O group can be represented by one of two symbols for a dipole—the arrow with the cross at the positive end or the pair of δ+ and δ– symbols.
То есть, орбитали кислорода ближе к связывающим МО по энергии, поэтому электронная плотность больше у кислорода, и он имеет частичный \delta^{-} заряд.
А теперь взглянем на диаграмму МО метана:
По той логике, так как АО водородов ближе к связывающим МО по энергии, то водороды имеют частичный \delta^{-} заряд. НО, по шкале Полинга, ЭО углерода больше, чем ЭО водорода. А я всю жизнь думал: какой атом электроотрицательнее, тот и \delta^{-}. Почему получаются разные результаты?
Моя догадка:
В книге ранее говорилось, что мы упростили картину, гибридизовав атомы, то есть соединили две разные теории (ВС и МО) для удобства. То есть, если не гибридизовав атомы, учесть все взаимодействия АО атомов и математическим путем вычислить все МО метана, то все заработает и АО углерода будут поближе к связывающим МО, чем АО водорода
Вроде бы нашел точную диаграмму МО метана:
Да, 2s углерода ближе к \sigma_{s}, но в других связывающих МО, АО водорода ближе. Кстати, тут АО водорода выше по энергии 2s углерода. А в диаграмме клейдена она ниже… Может клейден погнал, что поставил 1s H ниже чем 2s C? Тогда может sp3 будет пониже 1s H, и тем самым поближе к bonding MO?
Он кстати в моей последней диаграмме выше чем 2s углерода. Но вот именно, что если он ниже чем орбитали углерода, то он ближе к связывающим МО. А это значит, что он \delta^{-}
Как бы сложно сказать, что МО сильно смещена в сторону водородов. Я бы сказал она ± равномерно поделена. А \delta- берется (я предполагаю) за счет более низкой МО, которая больше сосредоточена на углероде.
Локализованные орбитали это норм, но рисовать их в учебных материалах для начинающих, причём называть “молекулярные орбитали” это прям выстрел в ногу для тех, кто захотел разобраться (они там делают предупреждение, но для начинающего это ничего не значит). Потом человек будет иметь странные концепции в голове и это может вырасти в целое дерево непонимания, и такой отравленный плод может много рассуждений поломать на годы вперед.
Именно поэтому всё и поломалось. Почему когда пользуемся чистым МО можем спокойно говорить “кто ближе/у кого коэффциенты больше” тот и электроны на себе держит? Потому что атомные орбитали из МО симметричные относительно ядра атома. А теперь нарисуй sp3 орбиталь углерода… она сама по себе уже дипольный момент несет, ведь не симметрична относительно ядра углерода и смещена в сторону водорода. А значит такая логика неприменима.
А вообще не понятно там минус учитывать и -16 будет ниже -13, или там без минуса и 16 выше 13. Я просто видел энергии с положительным значением, но кажется там просто можно обеими знаками брать